
Einzureichende Unterlagen
Anträge nach AMG reichen sie bitte über das CTIS-Portal der EMA ein.
Allgemeines
Das Medizinprodukte-Durchführungsgesetz (MPDG) ersetzt für Medizinprodukte, die unter die Verordnung (EU) 2017/745 fallen (MDR- Medical Device Regulation), mit seinem vollständigen Inkrafttreten am 26. Mai 2021 das Medizinproduktegesetz (MPG).
Definitionen
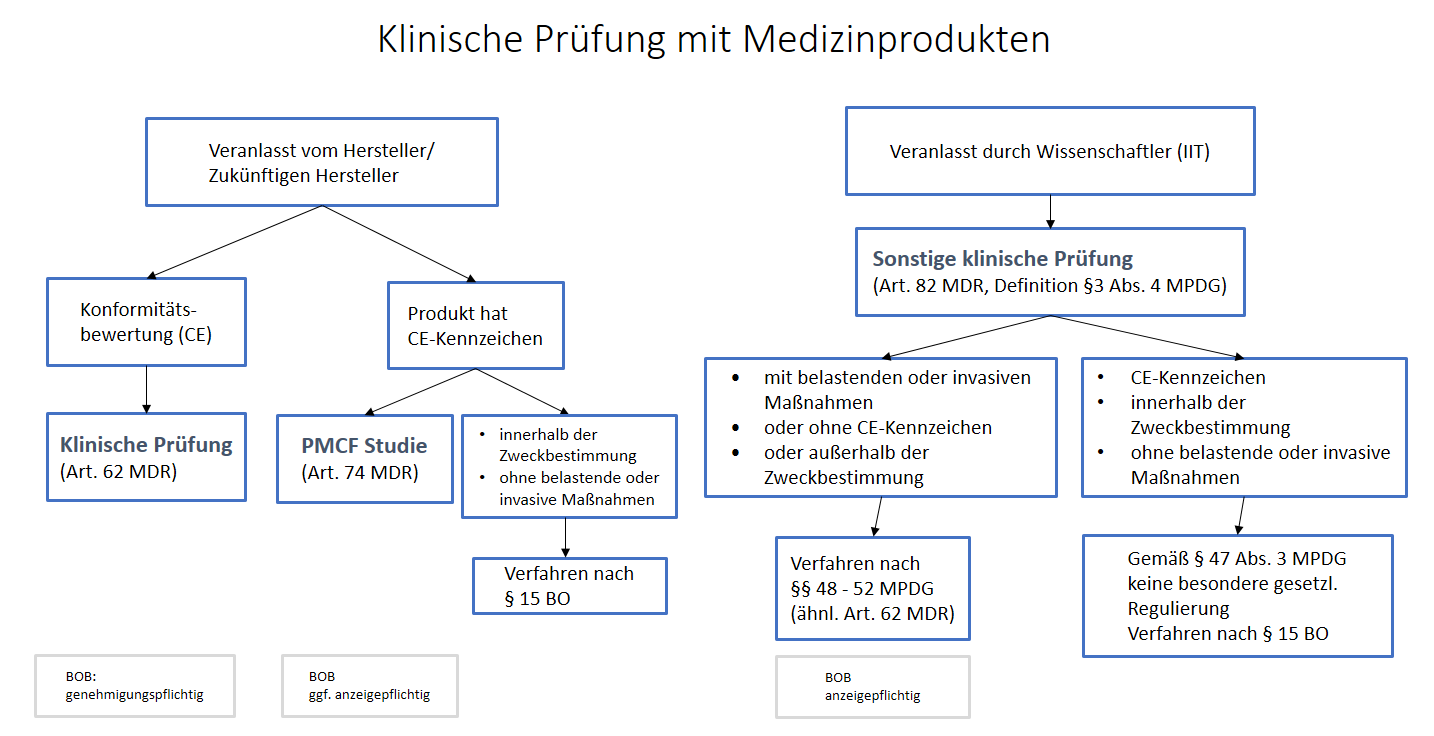
„klinische Prüfung“ (MDR Art. 2 Abs. 45)
bezeichnet eine systematische Untersuchung, bei der ein oder mehrere menschliche Prüfungsteilnehmer einbezogen sind und die zwecks Bewertung der Sicherheit oder Leistung eines Produkts durchgeführt wird
„sonstige klinische Prüfung“ (MPDG §3 Abs. 4)
eines Produktes ist eine klinische Prüfung, die
- nicht Teil eines systematischen und geplanten Prozesses zur Produktentwicklung oder der Produktbeobachtung eines gegenwärtigen oder künftigen Herstellers ist,
- nicht mit dem Ziel durchgeführt wird, die Konformität eines Produktes mit den Anforderungen der Verordnung (EU) 2017/745 nachzuweisen,
- der Beantwortung wissenschaftlicher oder anderer Fragestellungen dient und
- außerhalb eines klinischen Entwicklungsplans nach Anhang XIV Teil A Ziffer 1 Buchstabe a der Verordnung (EU) 2017/745 erfolgt;
Übergangsregelung
Nach Artikel 120 Absatz 11 der Verordnung (EU) 2017/745 dürfen klinische Prüfungen, die
– gemäß Artikel 10 der Richtlinie 90/385/EWG oder
– gemäß Artikel 15 der Richtlinie 93/42/EWG
vor dem 26. Mai 2021 eingeleitet wurden, weitergeführt werden.
Nach § 99 Absatz 3 MPDG gelten klinische Prüfungen als eingeleitet im Sinne der Verordnung, wenn für sie jeweils in der Zeit vom 20. März 2010 bis einschließlich 25. Mai 2021 die zuständige Ethik-Kommission eine zustimmende Bewertung und die zuständige Bundesoberbehörde eine Genehmigung oder eine Befreiung von der Genehmigungspflicht erteilt haben.
Änderungsanträge und Anzeigen nach §§ 20-24 MPG für entsprechend bereits begonnenen Studien können vom Sponsor über das internetbasierte Erfassungssystem des DMIDS gestellt werden.
Bewertung einer klinischen Prüfung eines Medizinprodukts gemäß Art. 62 MDR (Verordnung (EU) 2017/745)
Klinische Prüfungen haben gemäß den Bestimmungen nach Art. 63-80 zu erfolgen, wenn sie als Teil der klinischen Bewertung für Konformitätsbewertungszwecke zu einem oder mehreren der folgenden Zwecke durchgeführt werden:
- zur Feststellung und Überprüfung, dass ein Produkt so ausgelegt, hergestellt und verpackt ist, dass es unter normalen Verwendungsbedingungen für einen oder mehrere der in Art.2 Abs. 1 aufgelisteten spezifischen Zwecke geeignet ist und die von seinem Hersteller angegebene bezweckte Leistung erbringt;
- zur Feststellung und Überprüfung des von seinem Hersteller angegebenen klinischen Nutzens eines Produkts;
- zur Feststellung und Überprüfung der klinischen Sicherheit des Produkts und zur Bestimmung von bei normalen Verwendungsbedingungen gegebenenfalls auftretenden unerwünschten Nebenwirkungen des Produkts und zur Beurteilung, ob diese im Vergleich zu dem von dem Produkt erbrachten Nutzen vertretbare Risiken darstellen.
Unterlagen
dem Antrag sind die in Anhang XV Kapitel II MDR aufgeführten Unterlagen beizufügen
Bewertung einer klinischen Prüfung eines Medizinprodukts gemäß Art. 74 MDR (Verordnung (EU) 2017/745)
Klinische Prüfungen in Bezug auf Produkte, die die CE-Kennzeichnung tragen (auch „klinische Prüfung nach dem Inverkehrbringen“ bzw. PMCF – post-market clinical follow-up)
1. Weitergehende Bewertung eines Produkts, das bereits die CE-Kennzeichnung gemäß Artikel 20 Absatz 1 trägt und das im Rahmen seiner Zweckbestimmung angewendet wird, mit zusätzlichen invasiven oder belastenden Maßnahmen für die Prüfungsteilnehmer zu den bei normalen Verwendungsbedingungen durchgeführten Verfahren
Der Sponsor übermittelt die Unterlagen gemäß Anhang XV Kapitel II.
Für klinische Prüfungen nach dem Inverkehrbringen gelten Artikel 62 Absatz 4 Buchstaben b bis k und m, die Artikel 75 bis 77 und Artikel 80 Absatz 5 sowie die einschlägigen Bestimmungen des Anhangs XV.
2. Wird eine klinische Prüfung zur Bewertung eines Produkts durchgeführt, das bereits die CE-Kennzeichnung gemäß Artikel 20 Absatz 1 trägt, und außerhalb seiner Zweckbestimmung angewendet wird, so gelten die Artikel 62 bis 81.
Klinische Prüfungen, die nicht zu einem der in Artikel 62 Absatz 1 genannten Zwecke durchgeführt werden, müssen den Bestimmungen des Artikels 62 Absätze 2 und 3, Absatz 4 Buchstaben b, c, d, f, h und l und Absatz 6 genügen.
Die genauen Anforderungen um die Rechte, die Sicherheit, die Würde und das Wohl der Prüfungsteilnehmer zu schützen und die Einhaltung wissenschaftlicher und ethischer Grundsätze zu gewährleisten sind in §§48-52 MPDG festgelegt.
Der Antrag muss Folgendes enthalten:
Angaben und Unterlagen, die in Anhang XV Kapitel II der Verordnung (EU) 2017/745 genannt sind, mit Ausnahme der in Anhang XV Kapitel II Ziffer 1.5., 1.15., 3.1.1. und 4.2. der Verordnung (EU) 2017/745 genannten Angaben und Unterlagen
Bewertung einer sonstigen klinischen Prüfung eines Medizinprodukts gemäß Art. 82 MDR, §§48-52 MPDG
Klinische Prüfungen, die nicht zu einem der in Artikel 62 Absatz 1 genannten Zwecke durchgeführt werden, müssen den Bestimmungen des Artikels 62 Absätze 2 und 3, Absatz 4 Buchstaben b, c, d, f, h und l und Absatz 6 genügen.
Die genauen Anforderungen um die Rechte, die Sicherheit, die Würde und das Wohl der Prüfungsteilnehmer zu schützen und die Einhaltung wissenschaftlicher und ethischer Grundsätze zu gewährleisten sind in §§48-52 MPDG festgelegt.
Der Antrag muss Folgendes enthalten:
Angaben und Unterlagen, die in Anhang XV Kapitel II der Verordnung (EU) 2017/745 genannt sind, mit Ausnahme der in Anhang XV Kapitel II Ziffer 1.5., 1.15., 3.1.1. und 4.2. der Verordnung (EU) 2017/745 genannten Angaben und Unterlagen
Bewertung einer klinischen Prüfung eines in-vitro-Diagnostikums gemäß §§ 20-24 MPDG
Die Europäische Verordnung für In-vitro-Diagnostika (IVDR) trat gemeinsam mit der Verordnung für Medizinprodukte (MDR) am 25. Mai 2017 offiziell in Kraft. Die IVDR ist nach einer fünfjährigen Übergangszeit ab 26. Mai 2022 verpflichtend anzuwenden.
Die diesbezüglichen Anträge sowie weitere Anträge/Anzeigen, zum Beispiel bei Änderungen, werden bis dahin weiterhin nach §§ 20 – 24 MPG gestellt.
Gemäß § 3 Abs. 1 MPKPV ist der Antrag auf Genehmigung der zuständigen Bundesoberbehörde und auf zustimmende Bewertung der zuständigen Ethik-Kommission über das zentrale Erfassungssystem des DMIDS einzureichen.
Ein sonstiges Forschungsvorhaben ist ein Forschungsvorhaben, das weder dem Medizinproduktegesetz noch dem Arzneimittelgesetz unterliegt, jedoch biomedizinische Forschung am Menschen oder epidemiologische Forschung mit personenbezogenen Daten beinhaltet.
Nach § 15 Abs. 1 der Berufsordnung für Ärztinnen und Ärzte in Mecklenburg-Vorpommern muss sich ein Arzt „…vor der Durchführung biomedizinischer Forschung am Menschen – ausgenommen bei ausschließlich epidemiologischen Forschungsvorhaben – durch eine bei der Ärztekammer oder bei einer medizinischen Fakultät gebildeten Ethikkommission über die mit seinem Vorhaben verbundenen berufsethischen und berufsrechtlichen Fragen beraten lassen.“
Der Antrag an die Ethikkommission ist 2fach in Papierform und 1fach in elektronischer Form bei der Geschäftsstelle einzureichen.
Dem Antrag an die Ethikkommission ist unabhängig von der Art der klinischen Studie immer ein Studienprotokoll mit einer Synopse beizufügen. Die Synopsis des Studienprotokolls sollte knapp gehalten und in einer für Laien verständlichen Weise formuliert sein.
Patienteninformation und Einwilligungserklärung sollen getrennte Formulare sein
ebenso wie die Synopsis des Studienprotokolls sind die Patienteninformation und Einwilligungserklärung laienverständlich zu formulieren. Medizinische Fachbegriffe sind -wenn möglich- zu vermeiden, andernfalls sind die Begriffe zu erläutern.
Die Studienteilnehmer sind darüber zu informieren, dass ihre Teilnahme freiwillig ist und sie ohne Angabe von Gründen jederzeit ihre Einwilligung widerrufen können, ohne dass ihnen daraus Nachteile entstehen.
Der Passus zum Datenschutz, ist optisch hervorzuheben
Einzureichende Unterlagen: